Long QT Syndrome
Overview
Long QT syndrome (LQTS) is either an inherited (congenital) or acquired disorder of the electrical system of the heart that may result in sudden cardiac death (SCD) from episodes of ventricular tachycardia (VT). The LQTS is generally a result of disorders in the ionic sodium or potassium electrical channels that occupy the membranes of the cardiac muscle cells. This disorder in ionic channel conduction can lead to prolonged vulnerable periods in the cardiac cycle where extra-ventricular beats can lead to runs of ventricular tachycardia. There are at least 7 different genetic types of LQTS identified at this time, with LQT1, LQT2, and LQT3 being more common. It is estimated that congenital LQTS results in at least 3000-4000 sudden cardiac deaths in children each year with an estimated incidence of 1 out of every 10,000 – 20,000 people in the general population.
Acquired LQTS patients typically come to the attention of physicians after a secondary medical condition leads to symptomatic arrhythmias and a prolonged QT-interval on the 12-lead electrocardiogram. These conditions can include new medications, bradycardia (slow heart rates), and mineral deficiency, such as hypokalemia. The proposed mechanism is that these patients have a milder form of congenital LQTS, with a second trigger needed to expose the condition.
Diagnosis
The EKG finding of an inherited LQTS is typically a prolonged QT-interval on the 12-lead electrocardiogram. As discussed in the EKG section, the p-wave typically represents atrial activity, the QRS interval represents ventricular contraction, and the QT interval represents repolarization or recovery of the ventricular tissue so it can be stimulated again by the next p-wave. It is this prolongation of the QT-interval, or ventricular repolarization, that leads to the danger of spontaneous ventricular tachyarrhythmias, such as ventricular tachycardia. Not all patients have QT prolongation on their EKG at baseline; exercise treadmill tests and 24-hour ambulatory monitoring may help to clarify the diagnosis by capturing intermittent episodes. As well, a family history of sudden cardiac death or confirmed LQTS can be a clue to the diagnosis.
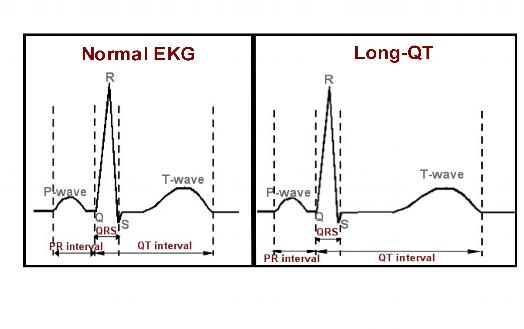
Figure 1. Representation of a normal EKG on the left with normal intervals labeled. The P-wave represents atrial activity, the QRS represents ventricular activation, and the QT interval represents ventricular recovery, or repolarization. Note the panel on the right, typical of a patient with Long-QT syndrome. The QT interval depicted here is significantly prolonged, with ventricular repolarization delayed, leading to an increased risk of ventricular tachyarrhythmias.
A particular stereotypical arrhythmia associated with patients with LQTS is a form of polymorphic VT known as “torsade de pointes”. It has a stricking visual appearance with a fast, wide-complexrhythm originating from the ventricles, with undulation or twisting about a baseline. The real danger and resultant sudden cardiac death occurs when this arrhythmia is sustained and degenerates into ventricular fibrillation with hemodynamic collapse.
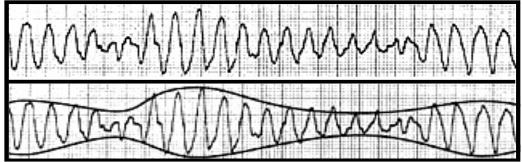
Figure 2. EKG strip of torsade de pointes (TdP) with a wide-complex ventricular tachycardia with alternating undulations, or twisting, about the baseline. This is a characteristic rhythm encountered in patients with long QT syndrome (LQTS).
The triggers for ventricular arrhythmias in patients with LQTS are diverse, but typically are initiated by triggers, such as loud noises (i.e., alarm clocks, telephone rings), exercise (i.e, swimming), and emotions.
Symptoms
Symptoms of LQTS include loss of consciousness (syncope), dizziness, and palpitations. The initial presentation can be fatal, with sudden cardiac death. Some patients may present with a history of episodic syncope for years. Any syncope in a young patient should prompt an aggressive work-up to include a baseline 12-lead EKG. However, an even larger percentage of patients may be asymptomatic and come to medical attention only after family members with the condition are diagnosed or a prolonged QT interval is identified on their resting EKG. A difficult aspect of the LQTS is that family members of an affected individual may have inherited the genetic disorder, but never or only mildly manifest any symptoms. This is called “penetrance”; that is, some family members may have a low penetrance with certain genetic LQTS with only mild to no symptoms, while others may first exhibit sudden cardiac death.
Prognosis
The risk for sudden cardiac death in LQTS is difficult to quantify in all patients. Clearly symptomatic patients and patients with aborted sudden cardiac death are at a high risk for recurrent events. There are certain genotypes of LQTS that have higher risks for sudden cardiac death in certain age groups. Males tend to have a higher initial event rate at younger ages than females, whereas females have a higher incidence into adulthood. As well, the greater the baseline prolonged QT-interval, the higher the risk of suffering a clinical event. The severity of the LQTS in an affected individual does not necessarily predict the future event rate of the siblings.
Treatment
The treatment of LQTS does differ between acquired and congenital forms. In the acquired forms, correction of mineral deficiencies, increasing the baseline heart rate, and withdrawal of offending drugs is critical to the therapy.
The treatment for congenital LQTS is more complicated and contentious. Generally, avoidance of QT prolonging agents and known environmental triggers are a necessity. Competitive sports are contraindicated in symptomatic LQTS, and discouraged in asymptomatic patients. Therapy with B-blockers is recommended for both symptomatic and asymptomatic LQTS patients, with an implantable cardioverter defibrillator (ICD) placed in patients with symptomatic recurrences on b-blockers or for secondary prevention in patients who have suffered a sudden cardiac death. A thorough discussion of your medical history and records is critical for your physician to tailor your therapy accordingly.
For further information on Long QT syndrome and a list of offending medications that prolong the QT interval, please consult the following websites:
- Arrhythmias
- Sick Sinus Syndrome
- Atrioventricular (AV) Block
- Atrial Tachycardia
- AV nodal reentrant tachycardia (AVNRT)
- Wolff-Parkinson White Syndrome (WPW) and Atrioventricular reciprocating tachycardia (AVRT)
- Atrial Flutter
- Atrial Fibrillation
- Cardioversion
- Ventricular Tachyarrhythmias
- Long QT Syndrome
- Sudden Cardiac Death (SCD)
- Syncope


 Silver Spring Office
Silver Spring Office  DC Office (at Providence Hospital)
DC Office (at Providence Hospital)  Hagerstown Office
Hagerstown Office